
(Khilji, Muhammad Faisal. (2013). klippel feil syndrome)
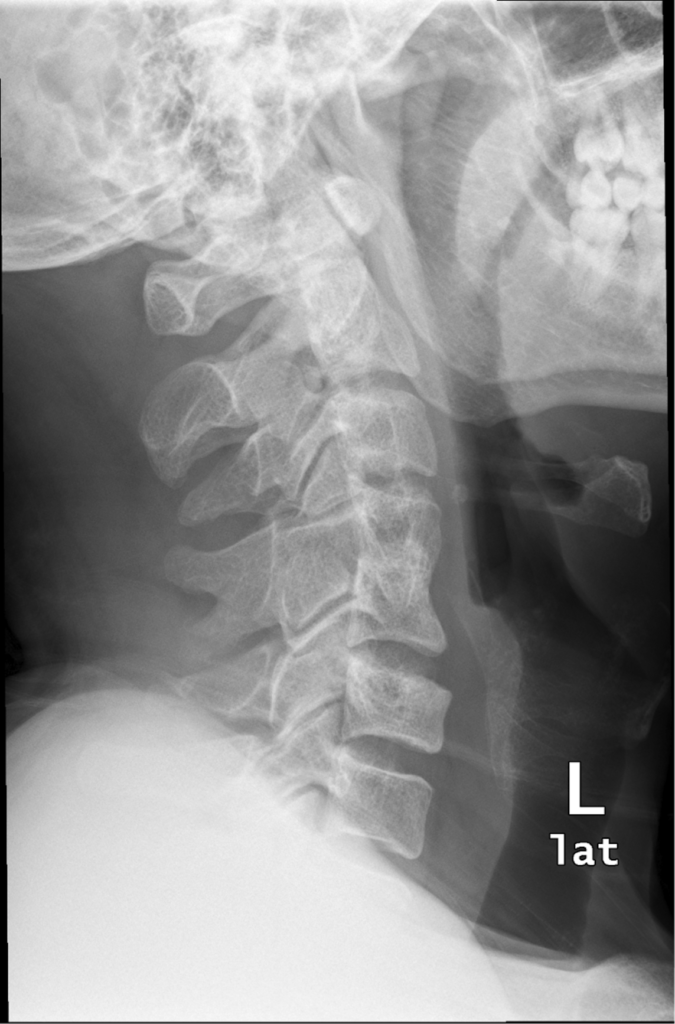
Zespół wad wrodzony, opisany po raz pierwszy przez Maurice Klippel i Andre Feil w 1912r, w którym dochodzi do fuzji (zrośnięcia) 2 lub więcej kręgów szyjnych.
Występowanie: 1/40-42 000 urodzeń. Współwystępowanie z zespołem Chiari : zespól Klippel- Feil współwystępuje od 3 do 5 % pacjentów z zespołem Chiari typu I.
Zakres fuzji kręgów obejmuje tylko trzony kręgów ( wrodzony blok kostny) ale także może dotyczyć również całych kręgów (nasady, stawy międzykręgowe, blaszki, wyrostki kolczyste).Powodem powstania jest zaburzenie prawidłowej segmentacji somitów (elementy prawidłowego embrionu), między 3 a 8 tygodniem ciąży
Charakterystyczna triada objawów ( występuje w 50 % przypadków).
- Obniżenie linii włosów na karku.
- Krótka szyja.
- Ograniczenie ruchomości głowy i szyi (może nie być ewidentne, gdy fuzja dotyczy mniej niż 3 kręgów szyjnych lub obejmuje jedynie dolny odcinek kręgosłupa szyjnego). Ograniczenie dotyczy raczej rotacji niż ruchu zgięcia-odgięcia.
Poza wyżej wymienionymi nieprawidłowościami dotyczącymi układu kostnego u pacjentów z zespołem Klippel-Feil współwystępować mogą również m.in. : skolioza, deformacja Sprengle’a (wrodzone uniesienie łopatki), wady serca i nerek, kręcz szyi, porażenie nerwu twarzowego.
Typy:
Typ I: obejmuje fuzje elementów wielu( >2) kręgów w jeden blok kostny.
Typ II: obejmuje fuzję tylko jednego lub dwóch kręgów szyjnych.
Typ III: Obejmuje typ I lub typ II wraz z wadami kręgosłupa piersiowego i/lub lędźwiowego.
Leczenie:
Preferuje się leczenie zachowawcze, z naciskiem na modyfikacje stylu życia z unikaniem urazów w obrębie kręgosłupa szyjnego ( wysoka predyspozycja do uszkodzenia rdzenia kręgowego). Leczenie, w przypadku pojawienia się zespółu bólowego polega na rehabilitacji oraz noszeniu kołnierza ortopedycznego.
W sytuacji stwierdzenia postępującej niestabilności kręgosłupa, rozważyć należy leczenie operacyjne z dekompresją kanału kręgowego (z lub bez stabilizacji).
lek. Artur Balasa